Большие гранулярные лимфоциты бгл

Лейкоз из больших гранулярных лимфоцитов (БГЛ) – диагностика, лечениеСиндром, характеризующийся пролиферацией больших гранулярных лимфоцитов (БГЛ), в качестве самостоятельной нозологической формы был впервые описан в 1977 г. На тот момент точного терминологического определения заболеванию дано не было. R. McKenna и соавт. описали хроническое лимфопролиферативное заболевание с необычными клиническими, морфологическими, ультраструктурными и иммунологическими характеристиками. За последние три десятилетия в литературе это заболевание называли по-разному: Ту-лимфопролиферативное заболевание, хронический Т-клеточный лимфоцитоз с нейтропенией, Т-клеточный хронический лимфолейкоз (Т-ХЛЛ), Т8+ Т-ХЛЛ. Все эти термины в той или иной степени отражали морфологические, иммунологические и функциональные характеристики опухолевых клеток. Так, в работе J. Brouet с соавт. лейкоз из больших гранулярных лимфоцитов охарактеризован как Т-клеточный хронический лимфолейкоз. Одиннадцать случаев, описанных авторами, как показал последующий анализ, включали двух пациентов с Т-клеточным пролимфоцитарным лейкозом и девять с лейкозом из больших гранулярных лимфоцитов. По современным представлениям, существование Т-клеточного хронического лимфолейкоза оспаривается. Лишь в немногих исследованиях он охарактеризован как отдельное заболевание. J. Hoyer и соавт., опираясь в основном на морфологические критерии, описали 25 случаев Т-ХЛЛ. Следует ли эти случаи относить к Т-ХЛЛ или мелкоклеточному варианту Т-клеточного пролимфоцитарного лейкоза (Т-ПЛЛ), в большей степени является предметом семантических, а не терминологических дискуссий. Е. Matutes и соавт., изучив электронно-микроскопические, цитогенетические, иммунологические и клинические особенности Т-ХЛЛ у этой группы больных, небезосновательно относят его к мелкоклеточному варианту Т-ПЛЛ. Таким образом, данные литературы, посвященной Т-клеточным лимфопролиферативным заболеваниям, следует интерпретировать с большой осторожностью, учитывая весь спектр новых методов обследования и современные классификационные подходы. Выделению лейкоза из больших гранулярных лимфоцитов в отдельную нозологическую форму немало способствовало изучение функции и морфологии различных популяций Т-лимфоцитов. Большие гранулярные лимфоциты составляют 5—20 % от общего количества лимфоцитов крови (в абсолютных значениях от 250 до 450 в 1 мкл крови). Это клетки диаметром 12—15 мкм, с умеренным или широким ободком цитоплазмы слабобазофильного цвета, содержащей нежные или более грубые (плотные) азурофильные гранулы, размер и количество которых значительно варьируют. Ядра клеток округлые или овальные, располагаются в центре или несколько эксцентрично. Хроматин ядер конденсированный, ядрышки не просматриваются. Соответственно двум типам больших гранулярных лимфоцитов выделяют 2 варианта лейкоза из БЛГ (ЛБГЛ): из NK-клеток (CD3-) и цитотоксических Т-клеток (CD3+). NK-ЛБГЛ встречается значительно реже, чем Т-ЛБГЛ, в основном в азиатском регионе, характеризуется агрессивным течением с поражением лимфатических узлов, печени, селезенки и наличием В-симптомов. Клиническая характеристика лейкоза из больших гранулярных лимфоцитов
Подавляющее большинство случаев лейкоза из больших гранулярных лимфоцитов представлено его Т-клеточным вариантом. Болеют в основном люди пожилого возраста, несколько чаще женщины. Т-клеточный лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) характеризуется доброкачественным клиническим течением и нередко протекает бессимптомно. Диагноз может быть поставлен при рутинном клинико-лабораторном обследовании. К диагностическим критериям Т-клеточного лейкоза из больших гранулярных лимфоцитов относятся увеличение абсолютного количества больших гранулярных лимфоцитов более 2 • 109/л с CD3+CD8+-иммунофенотипом в течение 6 мес и более, нейтропения и/или анемия. Известны случаи Т-клеточного лейкоза из больших гранулярных лимфоцитов с меньшим содержанием больших гранулярных лимфоцитов. У большинства пациентов симптомы заболевания связаны с рецидивирующими инфекциями на фоне нейтропении. Приблизительно у 20 % больных развитию Т-клеточного лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) предшествует длительный анамнез ревматоидного артрита. В этих случаях при физикальном обследовании определяется увеличение селезенки, обычно выступающей менее чем на 10 см из-под края реберной дуги. У половины больных обнаруживают спленомегалию, менее чем у 20 % пациентов манифестация заболевания проявляется кожными поражениями в виде папулезной сыпи. В противоположность В-ХЛЛ лимфаденопатия является достаточно редким симптомом (менее 5 % от всех случаев). В клиническом анализе крови определяется нормальное или незначительно повышенное количество лейкоцитов (5—30•109/л) с абсолютным лимфоцитозом (>5•109/л), причем клетки с характерными морфологическими особенностями больших гранулярных лимфоцитов. В подавляющем большинстве случаев количество нейтрофилов составляет менее 1,5•109/л. Анемия и тромбоцитопения выявляются приблизительно у 20 % больных. Опухолевые клетки при Т-клеточном лейкозе из больших гранулярных лимфоцитов имеют характеристики антигенактивированных цитотоксических Т-лимфоцитов, однако их антигенная специфичность точно не установлена. По данным некоторых исследователей, в сыворотке больных Т-клеточным лейкозом из больших гранулярных лимфоцитов обнаружены антитела к HTLV-I/II, но убедительных свидетельств клональной интеграции ретровируса в геном лейкозных клеток не получено. Одним из основных механизмов персистенции лимфоцитов при Т-клеточном лейкозе из больших гранулярных лимфоцитов является нарушение регуляции апоптоза. Нормальные цитотоксические Т-лимфоциты, распознавая вирусные пептиды при участии главного комплекса гистосовместимости, имеют высокий уровень экспресии Fas/CD95 и Fas/CD95-лиганда. Инфицированные вирусом клетки-мишени затем элиминируются путем Fas-зависимого апоптоза. Fas-лиганд относится к семейству белков фактора некроза опухоли и индуцирует апоптоз путем связывания со своим рецептором, который также известен как APO-I или CD95. В уничтожении антигенактивированных цитотоксических Т-лимфоцитов задействован тот же самый механизм. Показано, что лимфоциты при Т-ЛБГЛ также имеют высокий уровень экспрессии Fas и Fas-лиганда и их накопление обусловлено нарушением Fas-зависимого апоптоза. Устойчивая экспрессия и экскреция Fas-лиганда Т-БГЛ может являться одним из возможных механизмов развития нейтропении, особенно при отсутствии четкой корреляции между степенью нейтропении и объемом поражения костного мозга. В норме нейтрофилы экспрессируют CD95 и подвергаются Fas-зависимому апоптозу, поэтому можно предположить, что избыточная секреция Fas-лиганда опухолевыми БГЛ может быть одной из причин нейтропении при Т-клеточном лейкозе из больших гранулярных лимфоцитов (Т-ЛБГЛ). Важной отличительной чертой Т-клеточного лейкоза из больших гранулярных лимфоцитов (Т-ЛБГЛ) является ассоциация с другими заболеваниями. По данным ряда авторов, около 20—30 % больных Т-ЛБГЛ имеют серологические и клинические признаки ревматоидного артрита. – Также рекомендуем “Синдром Рихтера – история изучения” Оглавление темы “Лейкозы”:
|
Источник
Цереброретинальный ангиоматоз (болезнь Гиппеля-Линдау, БГЛ) – методы диагностики, лечения по Европейским рекомендациям
а) Синонимы. Болезнь Гиппеля-Линдау (БГЛ) также называется цереброретинальный ангиоматоз.
б) Эпидемиология. Болезнь Гиппеля-Линдау (БГЛ) является относительно редким нейрокожным синдромом, с частотой примерно 1:40000 новорожденных.
в) Этиология. Болезнь Гиппеля-Линдау (БГЛ) это аутосомно-доминантное наследственное заболевание с высокой пенетрантностью (70%) и варьирующей экспрессией. Причиной этого расстройства, как полагают, является вовлечение гена-супрессора опухоли пли линейное расположение генов на коротком плече хромосомы 3 (3р25-26).
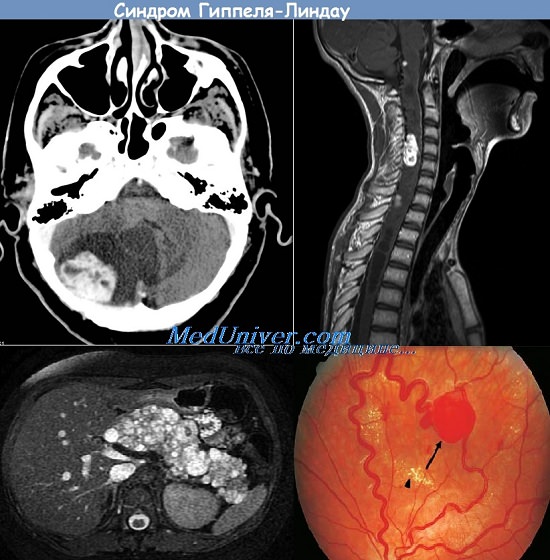
г) Глазные проявления цереброретинального ангиоматоза. Гемангиобластомы сетчатки: у 50% БГЛ пациентов. Эти повреждения часто периферические и многочисленные и, как правило, двусторонние. Диагностика облегчается за счет использования флюоресцентной ангиографии.
В связи с относительным риском кровоизлияния гемангиобластомы сетчатки следует рассматривать для лечения и у бессимптомных пациентов; основой лечения является лазерная терапия.
д) Неврологические проявления. Гемангиобластома (ГБ): наиболее частое явлениеу пациентов с БГЛ. 75% всех очагов расположено в мозжечке, повреждения спинного мозга и больших полушарий редки. Пациенты с мозжечковым поражением обычно имеют симптомы внутричерепной гипертензии (головная боль, рвота), которая в отдельных случаях может быть острой в связи с кровоизлиянием в опухоль.
МРТ обычно показывает гипоин-тенсивный в Т1-режиме и гиперинтенсивный в Т2-режиме узелок, связанный с кистозным компонентом. Узлы обычно примыкают к мягкой мозговой оболочке и хорошо накапливают контраст. Для небольших поражений наиболее чувствительным методом является ангиография. Лечение заключается в хирургическом удалении солидной части; стенку кисты не обязательно удалять полностью.
Спинальные гемангиоблстомы (ГБ) похожи на мозжечковые, они, как правило интрамедуллярные, хотя упираются в поверхность мягкой мозговой оболочки. Они могут быть ассоциированы с сирингомиелией. Хирургическое иссечение—метод выбора.
Другие необычные проявления, связанные с БГЛ, — киста поджелудочной железы, рак почек и феохромоцитома.
– Также рекомендуем “Энцефалотригеминальный ангиоматоз (синдром Стерджа-Вебера) – методы диагностики, лечения по Европейским рекомендациям”
Оглавление темы “Нейрокожные синдромы.”:
- Нейрофиброматоз 1 типа – методы диагностики, лечения по Европейским рекомендациям
- Нейрофиброматоз 2 типа – методы диагностики, лечения по Европейским рекомендациям
- Болезнь Бурневилля (туберозный склероз) – методы диагностики, лечения по Европейским рекомендациям
- Цереброретинальный ангиоматоз (болезнь Гиппеля-Линдау, БГЛ) – методы диагностики, лечения по Европейским рекомендациям
- Энцефалотригеминальный ангиоматоз (синдром Стерджа-Вебера) – методы диагностики, лечения по Европейским рекомендациям
- Наследственная геморрагическая телеангиэктазия (НГТ, болезнь Рандю-Вебера-Ослера) – методы диагностики, лечения по Европейским рекомендациям
Источник
Представлена клинико-гематологическая и иммунофенотипическая характеристика Т-клеточного варианта лейкоза из больших гранулосодержащих лимфоцитов. Выделены дифференциально-диагностические признаки и приведены протоколы лечения.
Синдром, характеризующийся пролиферацией больших гранулосодержащих лимфоцитов (БГЛ), был впервые описан в 1977 г. Опухолевая природа этого синдрома долгое время вызывала сомнения в связи с отсутствием надежных способов доказательства клональности. Проведение иммунофенотипических и молекулярно-генетических методов исследования для определения Т-клеточной клональности позволило охарактеризовать своеобразную лимфатическую опухоль и выделить ее среди многочисленных случаев Т-клеточного лимфоцитоза [1,2].
На протяжении последних лет в Украине наблюдается рост числа больных с хроническими лимфопролиферативными заболеваниями. Клинические проявления хронического лейкоза из больших гранулосодержащих лимфоцитов чаще всего сопровождаются гранулоцитопенией. Опухолевые клетки демонстрируют своеобразную морфологию, давшую название заболеванию. Характерен умеренный лимфатический лейкоцитоз, часто с абсолютной нейтропенией. Не существует единства мнений в вопросе о том, при каком количестве лимфоцитов может быть установлен диагноз лейкоза из больших гранулосодержащих лимфоцитов. Полагают, что уровень лимфоцитов менее 5х109/л свидетельствует о реактивном лимфоцитозе, а более 5х109/л указывает на наличие Т-клеточного лейкоза из БГЛ. В большинстве случаев при лейкозе из БГЛ при умеренном лимфоцитозе (~8х109/л) определяется более 2х109/л БГЛ [3].
Не выявлено также связи между выраженностью нейтропении и степенью поражения костного мозга. В связи с частым обнаружением в крови (по данным различных авторов) антинейтрофильных антител не исключена аутоиммунная природа нейтропении [4].
Выделение лейкоза из больших гранулосодержащих лимфоцитов в отдельную нозологическую форму немало способствовало изучению функции и морфологии Т-лимфоцитов. Иммунофенотипически БГЛ гетерогенны. 80-90% всех больших гранулосодержащих лимфоцитов являются естественными киллерами (NK-клетки). Гены Т-клеточного рецептора в этих клетках не перестроены. Они не экспрессируют на своей поверхности молекулы, ассоциированные с Т-клеточным рецептором (CD3, CD8, CD4), и имеют иммунофенотип CD56+, CD3-, CD8-, CD16+.
10-20% БГЛ – это цитотоксические Т-лимфоциты, имеющие иммунофенотип CD57+, CD3+, CD8+ и CD16+[1,3].
Многие аутоиммунные заболевания сопровождаются пролиферацией NK и Т-клеток (CD8+), особенно ревматоидный полиартрит, при этом зачастую развивается нейтропения различной степени выраженности и спленомегалия (синдром Фелти). Относительные и абсолютные лимфоцитозы могут быть следствием многих перенесенных заболеваний, в том числе инфекционных, как вирусной (аденовирусная инфекция, грипп, цитомегаловирусная инфекция, вирусные гепатиты), так и невирусной природы (токсоплазмоз, туберкулез, сифилис, малярия и др.). Как правило, эти лимфоцитозы поликлональны, содержание циркулирующих в крови лимфоцитов и их субпопуляционный состав нормализуется через 1-5 месяцев после болезни.
Иногда наблюдается моноклональный лимфоцитоз, дифференциальная диагностика которого может вызывать некоторые затруднения. Яркий пример – инфекционный мононуклеоз, при котором в периферической крови наблюдается абсолютный лимфоцитоз с преобладанием активированных Т-лимфоцитов с фенотипом Т-клеток – хелперов (CD3+CD4+) [4, 5].
Таким образом, дифференциальная диагностика хронических лейкозов из больших гранулосодержащих лимфоцитов и реактивных лимфоцитозов вызывает большие затруднения и невозможна без проведения иммунофенотипических исследований.
Результаты и обсуждение
В данной статье мы хотели бы рассмотреть вопросы диагностики и лечения хронического лимфолейкоза из больших гранулосодержащих лимфоцитовна примере клинического случая.
Больная П., 1947 года рождения, была направлена в гематологическое отделение в связи с анемией, лейкопенией и тромбоцитопенией на обследование. При осмотре не было выявлено увеличения лимфатических узлов, селезенки и печени. После проведения иммуноцитохимических исследований у больной не был установлен диагноз хронического лимфопролиферативного заболевания (не установлена моноклоновость процесса).
Больная находилась (на протяжении 4 лет) под наблюдением гематолога в поликлинике. В связи с низкими показателями количества лейкоцитов и эритроцитов, больная периодически принимала преднизолон. Частые воспалительные заболевания, что сопровождались повышением температуры тела до 38-39ºС, появление органомегалии (сплено- и гепатомегалии), а также изменения в анализе крови (лейкопения с лимфоцитозом) привели к вторичному обследованию больной в гематологическом отделении. На этот раз отмечалась бледность кожных покровов без геморрагических проявлений и увеличения лимфатических узлов. Обращала на себя внимание спленомегалия (на 5-6 см выступала из-под нижнего края реберной дуги) и гепатомегалия (на 2-3 см пальпировалась ниже реберной дуги).
В анализе крови: Hb 100г/л, эр. 2,95х1012/л, тр. 31,9х109/л, л. 2,5х109/л, с. 9%, лимф. 75%, мон. 11%, э. 4%, б. 1%, СОЭ 53мм/час. В миелограмме количество лимфоцитов составило 60%. Морфологически лимфоциты выглядели крупнее типичных малых лимфоцитов. Ядро имело круглую или овальную форму. По сравнению с сегментоядерным нейтрофилом хроматин менее конденсирован. Широкая светло-голубая цитоплазма, содержащая азурофильные гранулы. При проведении цитохимических исследований определялась яркая реакция на кислую фосфатазу и мелкогранулярная PAS-реакция в субстратных клетках. Иммунофенотипически мононуклеары периферической крови экспрессировали: CD3 80%, CD2 55%, CD8 65%, CD7 50%, CD4 22%, NK 59%, CD20 7%, CD19 5%.
На основании проведенных исследований, больной установлен диагноз хронического лимфолейкоза из больших гранулосодержащих лимфоцитов. После проведения 2-х курсов полихимиотерапии по схеме CHOP (циклофосфан, доксорубицин, винкристин, преднизолон) исчезла органомегалия и значительно улучшились показатели крови. Так, в анализе крови: эр. 4,0х1012/л, Hb 115 г/л, тр. 182,0х109/л, л. 4,2х109/л, п. 2%, с. 65%, лимф. 27%, мон. 4%, э. 2%, СОЭ 15 мм/час. В настоящее время больная наблюдается гематологом в поликлинике без лечения.
Данный случай полностью соответствует диагностическим критериям Т-клеточного лейкоза из больших гранулосодержащих лимфоцитов, к которому относится повышение абсолютного количества БГЛ, цитопения и иммунофенотипически преобладание в крови клеток, экспрессирующих CD3 и CD8.
В заключении отметим, что в настоящее время стандарты терапии больных с этой редкой патологией не разработаны. Показаниями для начала терапии являются нейтропения (менее 500 клеток/мкл), осложненная частыми инфекциями, глубокая анемия, выраженная спленомегалия. Медиана выживаемости при Т-клеточном варианте БГЛ превышает 10 лет. В связи с длительным хроническим течением заболевания оправдана выжидательная тактика, стремление, по возможности, избегать миелодепрессивной химиотерапии. В случае глубокой нейтропении применяют метотрексат в виде монотерапии или в сочетании с преднизолоном, циклоспорин, ростовые факторы [6].
По данным T.P. Loughran и соавт., монотерапия преднизолоном дает эффект только во время лечения: с уменьшением дозы уровень гранулоцитов снижается, а лимфоцитоз возрастает. Наконец, при выраженном прогрессировании заболевания применяют полихимиотерапию (СНОР), флюдарабин, метотрексат в сочетании с преднизолоном. Что касается диагностики Т-клеточного лейкоза из БГЛ требуется морфологическое изучение мазка периферической крови, а в трудных для диагностики случаях рекомендуется определение Т-клеточной клональности [7].
Выводы
- Диагностика Т-клеточного лейкоза из больших гранулосодержащих лимфоцитов должна включать иммуноцитохимические методы с использованием широкой панели моноклональных антител.
- Показаниями для назначения специфической терапии является нейтропения, которая осложняется частыми инфекциями, глубокой анемией и спленомегалией.
1Родионова И.А., 1Скрипниченко С.В., 2Булавина В.П.
1Национальный медицинский университет им. А.А. Богомольца,
2Киевская городская клиническая больница № 9
Литература
- Go R.S., Li C.Y., Tefferi A., Phyliky R.L. Acquired pure red cell aplasia associated with lymphoproliferative diseas of granular T lymphocytes. Blood 2006; 98:483-485.
- Jaffe E.S. The pathology of NK-cell lymohomas and leukemias. Exp Oncology 2007; 23:1-4.
- Morice W.G., Kurtin P.J., Leibson P.J. et al. Demonstration of aberrant T-cell and natural killer-cell antigen expression in all cases of granular lymphocytes leukaemia. Br J Hematol 2003; 120: 1026-1036.
- Mraz-Gemhard S., Natkunam Y., Hoppe R.T. et al. Natural killer/natural killer-like T-cell lymphoma, CD56+, presenting in the skin: an increasingly recognized entity with an aggressive course. J Clin Oncol 2007; 19: 2179-2188.
- Kothapalli R., Bailey R.D., Kusmartseva I. et al. Constitutive expression of cytotoxic proteases and down-regulation of proteases inhibitors in LGL leukaemia. Int J Oncol 2006; 22: 54-59.
- Shapiro M., Wasik M.A., Junkins-Hopkins J.M. et al. Complete remission in advanced blastic NK-cell lymphoma/leukaemia in elderly patients using the hyper-CVAD regimen. Hematol 2007; 74: 46-51.
- Loughran T.P., Lamy T. Current concepts: large granular lymphocyte leukaemia. Blood Rev 2003; 13: 230-240.
Источник