Апоптоз лимфоцитов при травме

Покоящиеся CD3+Т-лимфоциты для поддержания собственной жизнедеятельности требуют постоянного контакта с комплексами HLA — пептид аутоантигенов на поверхности клеток микроокружения. Кроме этого, выживать Т-лимфоцитам «помогают» цитокины (ИЛ-4, ИЛ-6, ИЛ-7 и ИЛ-12). Благодаря указанным влияниям неактивные лимфоциты характеризуются относительно высоким уровнем экспрессии антиапоптотического белка bcl-2 и низкой экспрессией молекул Fas и рецепторов к фактору некроза опухоли α. Таким образом, покоящиеся Т-клетки являются достаточно резистентными к запрограммированной гибели. Но стоит таким клеткам активироваться, как их готовность к апоптозу возрастает в десятки раз. Дело в том, что после специфической активации (с вовлечением антигенраспознающего рецептора) в цитоплазме Т-лимфоцитов резко снижается экспрессия белка bcl-2, а на мембране появляется значительное количество молекул Fas и рецепторов к ФНО-α. Описанное явление имеет глубокий биологический смысл. Активированная клетка не только приобретает способность к усиленному делению, но и становится более чувствительной к регуляторным влияниям, ограничивающим пролиферацию. С помощью этого механизма в организме поддерживается стабильное количество клеток и обеспечивается профилактика новообразований. Иными словами, любая активированная иммунокомпетентная клетка неминуемо гибнет, поскольку становится очень чувствительной к апоптозу. Реализация запрограммированной гибели осуществляется как с помощью взаимодействия Fas с FasL (мембран-ассоциированным или растворимым, отщепляемым от поверхности клеток-эффекторов металлопротеиназами), так и при действии ФНО-α, уровень которого значительно повышается практически при любом воспалении.
Возникает вопрос: если активированные Т-клетки гибнут, то каким образом осуществляется эффективный иммунный ответ? Дело в том, что при попадании чужеродных патогенов Т-лимфоциты получают дополнительные стимулы, временно отменяющие реализацию запрограммированной гибели. К ним относятся сигналы от факторов роста (например, ИЛ-2), адгезионных молекул и костимуляторов. Как только патоген уничтожается, Т-клетки перестают получать подобные сигналы и погибают путем апоптоза. Именно таким образом восстанавливается нормальное количество лейкоцитов после перенесенной инфекции.
Роль интерлейкина-2 в апоптозе лимфоцитов
Интерлейкин-2 является фактором роста Т-лимфоцитов. Благодаря этой функции цитокин должен усиливать апоптотическую гибель Т-клеток, так как повышает частоту их деления. Фактически же выходит, что при адекватной антигенной нагрузке интерлейкин-2, наоборот, повышает стойкость лимфоцитов к запрограммированной гибели. Сегодня этот феномен объясняют теорией обратной связи в контроле апоптоза Т-клеток. Согласно этой теории интерлейкин-2 стартово повышает чувствительность Т-лимфоцитов к апоптозу. Дальнейшая судьба клетки зависит от уровня антигенного и костимулирующего воздействия. В случае отсутствия антигенной стимуляции уровень секретируемого интерлейкина-2 снижается, в результате чего Т-клетки подвергаются «пассивному» апоптозу, связанному с нехваткой указанного цитокина. Он обеспечивается митохондриальными механизмами запрограммированной гибели. Этот путь является нормальным механизмом ограничения иммунного ответа после гибели патогена. В случае избытка антигенного воздействия имеет место «активный» апоптоз Т-лимфоцитов. Дело в том, что чрезмерная стимуляция антигенраспознающего рецептора сопровождается такой же чрезмерной экспрессией рецепторов апоптоза (молекулы Fas и рецептора к ФНО-а). Поэтому пролиферативное действие ИЛ-2 не в состоянии перекрыть проапоптотические сигналы, поступающие в Т-клетку по указанным рецепторам. Механизм «активного» апоптоза обеспечивает ограничение гиперактивации антигенспецифического клона эффекторных клеток в случае массового представления антигена в организме (например, при аутоиммунных реакциях, негативной селекции Т-клеток тимуса, реакции трансплантат против хозяина). Материал с сайта https://wiki-med.com
Чувствительность лимфоцитов к апоптозу
Чувствительность лимфоцитов разных классов к апоптозу существенно отличается. Так, Т-клетки более чувствительны к апоптозу, нежели В-лимфоциты. Это объясняется следующим образом: активация рецептора антигенного распознавания Т-лимфоцита приводит к резкому повышению чувствительности клетки к запрограммированной гибели; в то же время активация антигенраспознающего рецептора В-клеток обуславливает резистентность этих лимфоцитов к апоптозу. Указанные особенности имеют огромное значение в иммунном ответе. В организме существует большое количество аутореактивных лимфоцитов, способных распознавать собственные антигены. Но аутоиммунные реакции не развиваются, поскольку Т-клетки, активировавшиеся после распознавания аутоантигена, гибнут путем апоптоза. Хотя активированные В-лимфоциты способны выживать после взаимодействия со специфическим аутоантигеном, они не в состоянии развивать иммунный ответ без поддержки со стороны Т-хелперов.
Т-хелперы 1-го типа более чувствительны к апоптозу, нежели Т-хелперы 2-го. Эта особенность также имеет свое объяснение. Поскольку Th 1 способствуют развитию эффекторных механизмов, повреждающих собственные клетки (например, вирус-инфицированные или опухолевые), необходим мощный регуляторный механизм, который ограничивал бы самоповреждение при гиперактивации указанных лимфоцитов.
 На этой странице материал по темам:
На этой странице материал по темам:
что такое апоптоз зрелых лимфоцитов
апоптоз иммунология
активация апоптоза лимфоцитов
аптоз в лимфоцитов
что такое апоптоз лимфоцитов
Источник
Роль нарушения апоптоза в развитии злокачественных лимфом кожи.В последние годы внимание исследователей привлечено к изучению роли нарушений механизмов апоптоза в развитии злокачественной пролиферации клеток. Апоптоз — форма программированной физиологической гибели клетки, являющейся результатом реализации ее генетической программы. Механизм апоптоза довольно сложен, в нем участвуют система цитокинов, молекулярные межклеточные взаимодействия, гормоны, ионы Са2+, причем в зависимости от ситуации один и тот же фактор может как индуцировать, так и подавлять развитие апоптоза. Апоптоз опосредует удаление пула нефункционируюших незрелых лимфоцитов, то есть является эффективным способом удаления клеток с генетическими повреждениями. При подавлении апоптоза создаются условия для пролиферации клеток. Белок р53, который активно участвует в механизме апоптоза, является онкосупрессором, поскольку трансформирует сигнал о нерепарированных разрывах цепей ДНК в сигнал к развитию апоптоза. Иными словами, присутствие р53 приводит к гибели клеток с нарушениями в геноме. Мутации гена р53 нарушают механизм апоптоза, что приводит к выживанию таких клеток, и они становятся потенциальным источником опухолевого роста. В норме белок р53 не экспрессируется. его экспрессия индуцируется облучением, действием противоопухолевых цитостатических препаратов и другими агентами. В качестве регулятора апоптоза, «супрессора клеточной смерти», выступают белки семейства Вс1-2 — продукты протоонкогена Вс1-2. Все гемопоэтические илимфоидные клетки содержат белок Вс1-2. Экспрессия гена Вс1-2 регулируется в процессе развития лимфоцитов. Гиперэкспрессия Вс1-2 ограничивает апоптоз, что может косвенно способствовать пролиферации клетки. Так, гиперэкспрессия Вс1-2 вследствие транслокации его гена в локус иммуноглобулинов сопряжена с развитием фолликулярной В-кле-точнойлимфомы. Повышенную экспрессию р53 на лимфоцитах определяют у больных злокачественных лимфом кожи с наиболее тяжелым клиническим течением, в частности при таком морфологическом варианте, как крупноклеточная лимфома. Причем на опухолевых клетках узловатых очагов поражения экспрессия этого белка определяется чаще, чем на клетках очагов пятнисто-бляшечного характера. По экспрессии лимфоцитами р53 и Вс1-2 предлагают различать доброкачественные и злокачественные лим-фопролиферативные процессы в коже, а также прогнозировать исход заболевания. В то же время экспрессия маркеров апоптоза не всегда коррелирует с клинической тяжестью течения заболевания и трансформацией его в наиболее злокачественные морфологические варианты. Диагностическая и прогностическая значимость маркеров, отражающих апоптоз клеток, в клинической дерматологической практике требует дальнейших исследований. Таким образом, с помощью моноклональных антител удается определить иммунофенотипический и генотипический профиль пролиферирующих лимфоцитов, то есть не только установить их принадлежность к определенной популяции и субпопуляции, но и охарактеризовать тип опухоли и степень опухолевой прогрессии. Основные из этих маркеров названы ниже.
Характеристика рецепторов, типичных для лимфоцитов злокачественного клона у больных злокачественными лимфомами кожи1. Аберрантная экспрессия (коэкспрессия рецепторов Т- и В-лимфоцитов и/или Т-хелперов и Т-супрессоров. нулевые лимфоциты без Т- и В-рецепторов). Таким образом, в основе развитияу больных злокачественными лимфомами кожи лежит злокачественная пролиферация лимфоцитов, которые могут различаться по своим иммунофенотипическим и генотипическим характеристикам, что находит отражение в разнообразии клинических и морфологических вариантов ЗЛК и нередко связано с прогнозом для больных. – Также рекомендуем “Классификация злокачественных лимфом кожи.” Оглавление темы “Злокачественные лимфомы кожи.”: |
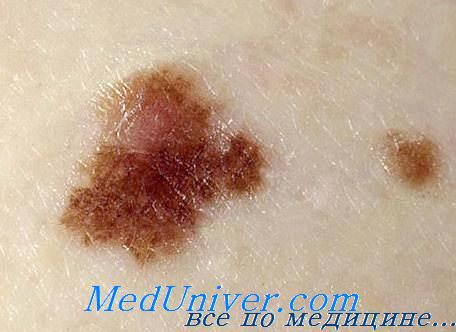
Источник
Т-лимфоциты и их субпопуляции
Тяжелая травма, ожоги, шок, обширные хирургические вмешательства сопровождаются разнообразными нарушениям специфического (табл. 55.1) и неспецифического (табл. 55.2) иммунитета. Методом проточной цитофлюориметрии в крови обнаруживают снижение числа Т-лимфо-цитов (CD3) и повышение числа моноцитов (CDI4). Кроме того, резко падает число Т-хелперов (CD4), уменьшается экспрессия рецептора ИЛ-2 на поверхности лимфоцитов. В то же время содержание цитоток-сических Т-лимфоцитов и Т-супрессоров (CD8), играющих основную роль в патогенезе посттравматической иммуносупрессии, не меняется или возрастает. В результате отношение лимфоцитов CD4/CD8 становится меньше единицы. Эти изменения сохраняются в течение 3 нед после травмы. В значительной степени подавляется индуцированная ми-тогенами пролиферация лимфоцитов в культуре, по-видимому, вследствие избыточной продукции проогагландина Е2.
Нарушения специфического иммунитета после травмы
- Угнетение кроветворения
- Лимфопения, отношение лимфоцитов CD4/CD8 < 1
- Угнетение пролиферации Т- и В-лимфоцитов
- Снижение секреции лимфокинов (ИЛ-2, ИЛ-3, интерферона у)
- Усиление секреции ИЛ-4 и ИЛ-10 лимфоцитами
- Снижение экспрессии рецепторов ИЛ-2
- Нарушение взаимодействия макрофагов с Т-лимфоцитами
- Снижение экспрессии антигена HLA-DR
- Отрицательные кожные пробы, основанные на аллергических реакциях замедленного типа
- Подавление дифференцировки и Лимфоцитов
- Подавленна синтеза и секреции IgM плазматическими клетками
- Переключение синте ia классов иммуноглобулинов с IgG не IgM
Нарушения неспецифического иммунитета после травмы
- Моноцнтоз
- Усиление секреции ФНОа и ИЛ -6 макрофагами и повышение уровней ФНОа и ИЛ-6 в сыворотке
- Угнетение секреции 11Л -1 макрофагами
- Усиление синтеза простагландина Е2 макрофагами и повышение его уровня в сыворотке
- Нарушения функций нейтрофилов Угнетение хемотаксиса Угнетение фагоцитарной активности Снижение экспрессии р2-интегринов Снижение синтеза лейкотриена В4
- Снижение, а затем повышение образования активных форм кислорода
- Увеличение активности эластазы в сыворотке Усиление синтеза белков острой фазы воспаления Повышение уровня СЗа в сыворотке
- Повышение активности миелопероксидазы, уровней катепсина, лактоферрина и неоптерина в сыворотке
- Дефицит фибронектина, других опсонинов, антитромбина III Снижение активности NK-лимфоцитов
Клинические исследования показали, что угнетение пролиферации лимфоцитов при тяжелой травме способствует развитию инфекционных осложнений. Кроме того, сразу после травмы наблюдается стойкое снижение синтеза ИЛ-2, что тоже играет важную роль в развитии иммунологических нарушений, т. к. ИЛ-2 в норме индуцирует пролиферацию Т-лимфоцитов и усиливает продукцию антител В-лимфоци-тами. Описано также подавление продукции ИЛ-3 и интерферона у лимфоцитами. Падает экспрессия HLA-DR (антигена HLA класса II) макрофагами, особенно при сепсисе. Добавление интерферона у к культуре лимфоцитов в таких случаях приводит к повышению экспрессии HLA-DR. Предполагают, что при травме нарушается взаимодействие Т-лимфоцитов с макрофагами, в результате чего страдает регуляция иммунного ответа и в значительной степени подавляются функции Т-лимфоцитов.
Источник
Нарушение апоптоза клеток при хроническом лимфолейкозе (ХЛЛ)
В патогенезе хронического лимфолейкоза большое значение всегда придавали нарушениям процесса программированной клеточной смерти — апоптоза. Лабораторные данные о кинетике патологических лимфоцитов показывают, что около 99 % лимфоцитов при хроническом лимфолейкозе находятся в фазе G0.
Период полужизни нормальных В-лимфоцитов составляет 5—7 дней, а патологические лимфоциты при хроническом лимфолейкозе существуют в течение многих месяцев. Лейкемические лимфоциты при хроническом лимфолейкозе обычно не экспрессируют или очень слабо экспрессируют Fas-антиген — трансмембранный гликопротеин, участвующий в регуляции апоптоза.
Стадия инициации апоптоза клеток
В связи с этим хронический лимфолейкоз долго рассматривался как болезнь, при которой лейкемические клетки инертны по отношению к сигналам пролиферации и делятся очень медленно, а огромная масса опухолевых клеток образуется не за счет их активной пролиферации, а в результате увеличения периода жизни и в связи с этим постепенного их накопления и вытеснения нормальных клонов.
В последние годы получены данные, способные изменить эти представления. Пока не было нетоксичных для живого организма веществ, способных включаться в структуру ДНК, все исследования кинетики делались на культуре лимфоцитов. В настоящее время появились нерадиоактивные изотопы, которые в организме человека могут включаться в структуру ДНК вновь образующихся клеток. Это позволило изучать кинетику лимфоцитов в организме больных хроническим лимфолейкозом.
Стадия программирования апоптоза клеток
У большинства пациентов метка была обнаружена в популяции лимфоцитов CD19+ CD5+ через 2 нед. Дальнейшие исследования показали, что у разных пациентов от 1 до 10 % патологических клеток заменяются каждую неделю. Это свидетельствует о довольно активных процессах как пролиферации, так и апоптоза.
Приведенные данные вполне согласуются с имеющимися лабораторными результатами, которые казались плохо объяснимыми, поскольку демонстрировали, что в культуре лимфоциты больных хроническим лимфолейкозом быстро подвергаются спонтанному апоптозу.
– Также рекомендуем “Влияние экспрессии гена BCL-2 и белка p27 на течение и прогноз хронического лимфолейкоза (ХЛЛ)”
Оглавление темы “Клиника хронического лимфолейкоза (ХЛЛ)”:
- Влияние мутации гена ТР53 на течение и прогноз хронического лимфолейкоза (ХЛЛ)
- Нарушение апоптоза клеток при хроническом лимфолейкозе (ХЛЛ)
- Влияние экспрессии гена BCL-2 и белка p27 на течение и прогноз хронического лимфолейкоза (ХЛЛ)
- Влияние b2-микроглобулина (b2М) на течение и прогноз хронического лимфолейкоза (ХЛЛ)
- Влияние антигена CD23 на течение и прогноз хронического лимфолейкоза (ХЛЛ)
- Влияние тимидинкиназы и лактатдегидрогеназы (ЛДГ) на течение и прогноз хронического лимфолейкоза (ХЛЛ)
- Сипмтомы хронического лимфолейкоза (ХЛЛ) – анализы
- Аутоиммунные осложнения при хроническом лимфолейкозе – аутоиммунная гемолитическая анемия (АИГА)
- Инфекционные осложнения при хроническом лимфолейкозе – причины
- Опухоли при хроническом лимфолейкозе – частота, причины
Источник
Апоптоз играет важную роль при становлении и функционировании иммунной системы. Эта форма гибели клеток сопровождает развитие клеток иммунной системы, особенно лимфоцитов, на этапе их селекции. С помощью механизма апоптоза происходит гибель клеток-мишеней кил- лерных лимфоцитов. При дальнейшем изложении материала апоптоз будет неоднократно упоминаться в связи с самыми разнообразными иммунологическими процессами. Именно потому, что апоптоз широко распространен при развитии лимфоцитов в центральных лимфоидных органах, этот процесс рассматривается в настоящем разделе.
Выделяют 2 основных варианта гибели клеток — некроз и апоптоз (табл. 3.19). Гибель, вызванная прямым повреждением или разрушением клеточной мембраны, называют некрозом. Другой вариант гибели клеток — апоптоз (от греч. алолтостгст — опадание листьев) или программированная гибель — реализуется в ответ на действие физиологических сигналов или в процессе срабатывания генетической программы клетки и служит необходимым условием существования многоклеточных организмов. Понятие «апоптоз» ввели в 1972 г. Дж. Керр (J.F. Kerr) и соавт. в связи с описанием формы гибели лимфоцитов при действии глюкокортикоидов.
Таблица 3.19. Сравнительная характеристика апоптоза и некроза клеток
| Показатель | Апоптоз | Некроз |
| Пусковой фактор | Повышение проницаемости мембран митохондрий или сигнал, воспринимаемый мембранными рецепторами | Неадекватные условия среды, токсические агенты |
| Скорость развития | 1-4 ч | lt; 1 ч |
| Причины гибели клетки | Нарушение функционирования энергетической системы клетки, деградация ДНК | Нарушение целостности мембраны, осмотические процессы |
| Изменение размера клетки | Уменьшение (сморщивание) | Увеличение (набухание) |
| Изменения ядра | Конденсация хроматина, пикноз, фрагментация | Набухание |
Окончание табл. 3.19
| Показатель | Апоптоз | Некроз |
| Изменения в цитоплазме | Конденсация цитоплазмы, уплотнение гранул | Лизис гранул |
| Изменения клеточной мембраны | Потеря микроворсинок, образование вздутий, уплотнение | Нарушение целостности |
| Состояние ДНК | Упорядоченная (межнук- леосомная) деградация | Неупорядоченная деградация |
| Энергозависимость | Зависит | Не зависит |
| Зависимость от синтеза макромолекул | Часто зависит | Не зависит |
| Примеры проявления | Гибель клеток при метаморфозе, отрицательной селекции лимфоцитов, гормонозависимой атрофии, интерфазной радиационной гибели лимфоцитов, гибели клеток-мишеней киллерных клеток | Гибель клеток от гипоксии, действия токсинов, вирусном цитолизе, комплементзависимом цитолизе |
Некроз происходит при нарушении целостности клеточной мембраны и проявляется набуханием клетки, лизисом гранул и ядра, неупорядоченной деградацией ДНК. Проявлениями апоптоза служат, наоборот, сморщивание клетки, уменьшение ее размеров, уплотнение наружной и внутриклеточных мембран (при сохранении их целостности) с деполяризацией, утратой микроворсинок и формированием вздутий, а также уменьшение размеров, уплотнение и фрагментация ядра. При этом происходят разрывы ДНК между нуклеосомами, в результате чего образуются фрагменты протяженностью, кратной 180—190 пар оснований (размер нуклеосомы). От клетки, подвергшейся апоптозу, отшнуровываются «апоптотические тельца» — фрагменты ядра, окруженные мембраной. Уже в процессе апоптоза гибнущие клетки подвергаются фагоцитозу. В результате продукты распада клеток не поступают в межклеточное пространство и не вызывают воспалительной реакции, которая обычно сопутствует некрозу клеток. Апоптоз развивается более медленно (1 ч и более), чем некроз (практически сразу после действия повреждающих факторов).
Механизмы апоптоза детально изучены на клетках нематоды Caenorhabditis elegans, у которой было идентифицировано 14 генов, контролирующих 4 фазы развития апоптоза. Обнаруженные у млекопитающих гомологи некоторых из этих генов также участвуют в развитии апоптоза. Выделяют 3 фазы развития апоптоза — включение пусковых механизмов, активацию каспаз и реализацию гибели. Апоптоз может быть запущен по двум механизмам — рецепторному и митохондриальному (табл. 3.20; рис. 3.65, 3.66).


Рис. 3.66. Митохондриальный механизм запуска апоптоза
Рецепторный механизм запуска апоптоза реализуется с участием мебран- ных рецепторных молекул, цитоплазматическая часть которых представлена доменом смерти (death domain), содержащим около 80 остатков. Эти молекулы относят к семейству рецепторов TNFa. Известно 6 таких рецепторов: Fas-рецептор (АРО-1, CD95, DR2), TNF-R1 (р55, CD120а, DR1), DR3, DR4, DR5, DR6. Их лиганды — Fas-лиганд (FasL, CD178 — для Fas-рецептора), цитокин TNFa (для TNFR1), TRAIL (TNF-related apoptosis-inducing ligand — для DR4 и DR5), TL1A (для DR3 и DR6) (табл. 3.20). Все лиганды организованы в виде тримеров. Их взаимодействие с рецепторами приводит к тримеризации последних, что запукает сигнальный каскад. При этом домены смерти приобретают способность взаимодействовать с аналогичными доменами адапторных белков FADD (Fas-associated death domain) и TRADD (TNF-receptor death domain). FADD распознает домены смерти в составе про- каспазы 8 и, взаимодействуя с ними, вызывает активацию каспазы 8 (см. далее). Результат действия TRADD аналогичен, но он реализуется посредством FADD. Формирующиеся в результате указанных взаимодействий молекулярные комплексы называют DISC (Death-inducing signaling complex).
Митохондриальный механизм запуска апоптоза реализуется при повреждении функций митохондрий, приводящем к нарушению проницаемости их мембраны. Решающую роль в этом пути запуска апоптоза играют белки семейства Bcl-2. Их разделяют на проапототические (Bid, Bax, Bak, Bcl-XS и др.) и антиапоптотические (Bcl-2, Bcl-XL, Mcl-1 и др.). Запуск сигналов к апоптозу связан с проапоптотическими белками, содержащими 1 домен ВН (Bcl-2 homology) — BH3. Белки этой группы блокируют анти-
апоптотические факторы типа Bcl-2, образуя с ними димеры. Кроме того, в результате олигомеризации Вах и Bak они формируют трансмембранные поры. В норме олигомеризация этих факторов подавляется антиапоптоти- ческими факторами. Через поры в мембране митохондрий в цитозоль выходят цитохром с и фактор Apaf-1 (Apoptose protease activation factor 1). Аpaf-1 и цитохром с в присутствии АТФ образуют комплекс с неактивной каспазой — прокаспазой 9. Этот комплекс называют апоптосомой. В ней происходит активация каспазы 9.
Рецепторный механизм апоптоза может быть прерван активацией ингибиторов каспазы 8. Митохондриальный механизм блокируется антиапопто- тическими факторами Bcl-2 и Bcl-XL связывающими проапоптотические факторы. Пути запуска апоптоза не являются изолированными. Так, рецепторный механизм приводит к активации митохондриального фактора Bid, что обусловливает подключение митохондриального механизма апоптоза.
Необходимо упомянуть также о механизме контроля за балансом пролиферации и апоптоза, осуществляемого метаболитами сфингомиелина. Из них роль проапоптотического фактора играет церамид, действующий через механизмы митохондриального (через фактор Bax) и рецепторного (через Fas-рецептор) путей.

Оба пути запуска апоптоза приводят к активации каспаз (рис. 3.67). Каспазы — группа цистеиных протеаз, расщепляющих полипептидную
связь после остатков аспарагиновой кислоты. Как уже отмечалось, рецепторный путь приводит к активации каспазы 8, митохондриальный — к активации каспазы 9. Эти ферменты относят к группе инициаторных каспаз. Их активация — результат агрегации вследствие взаимодействия с адаптерными белками (FADD, Apaf-1). При агрегации происходит аутокаталитическое отщепление длинного N-концевого участка каспазы с последующим формированием активного гетеродимера. После активации инициаторных каспаз процесс апоптоза становится необратимым.
Инициаторные каспазы вызывают частичный протеолиз (отщепление короткого продомена) и вследствие этого активацию исполнительных, или эффекторных каспаз — каспазы 3, реже — каспазы 6 и каспазы 7. Известно до 280 молекул-мишеней исполнительных каспаз, локализованных преимущественно в ядре. Расщепление молекул-мишеней определяет все проявления апоптоза. Действие каспаз на фактор ретинобластомы (Rb) и 5-изоформу протеинкиназы С обусловливает нарушение контроля клеточного цикла. Расщепление киназ МЕКК-1 и FAK приводит к изменениям, вызывающим ослабление адгезионной способности клетки, а расщепление гельсолина и киназы РАК определяет характерные изменения клеточной морфологии. Расщепление той же каспазой ядерных ферментов PARP (Poly-ADP-ribose polymerase — поли-АДФ-рибоза полимераза), а также ДНК-зависимой протеинкиназы нарушает процесс репарации ДНК. Одна из главных мишеней каспазы 3 — нейтральная эндонуклеаза CAD (Caspase- activated DNase), ответственная за межнуклеосомную фрагментацию ДНК в апоптотических клетках. Показана причинная связь гибели клетки с фрагментацией ДНК: введение в клетку гена, кодирующего активную форму CAD, вызывают ее гибель. Среди других причин апоптотической гибели клетки называют исчерпание ее энергетических ресурсов вследствие нарушения функций митохондрий и неконтролируемых расходов энергии на репарацию ДНК.
Фагоцитозу апоптотических клеток способствует экспрессия на их поверхности молекул, служащих для фагоцитов источником сигналов типа «съешь меня». Так, при апоптозе нарушается асимметрия мембраны, и фосфатидилсерин, в норме локализующийся на внутренней поверхности мембраны, оказывается экспонированным снаружи (выявление его экспрессии по связыванию с меченным аннексином V используют для идентификации апоптотических клеток). Появляющиеся на поверхности фосфатидилсерин, а также тромбоспондин и десиалированные остатки мембранных гликоконъюгатов распознаются рецепторами фагоцитов (как профессиональных, так и факультативных), что обеспечивает быстрый фагоцитоз апоптотических клеток. Такое завершение апоптоза чрезвычайно важно для организма, поскольку предотвращает поступление внутриклеточных компонентов, включая ДНК, в межклеточное пространство и последующее развитие воспаления и аутоиммунных процессов. В то же время чрезмерно интенсивное поглощение фрагментов ДНК может активировать (через внутриклеточные TLR) патологические процессы, ведущие к развитию системной аутоиммунной патологии, например, системной красной волчанки (СКВ).
Апоптозу принадлежит важная роль не только в селекции лимфоцитов, но и в других процессах, связанных с развитием лимфоцитов, морфогенезом лимфоидных органов, а также проявлением активности клеток иммунной системы, прежде всего в контактном цитолизе (табл. 3.21).
Таблица 3.21. Участие апоптоза в формировании иммунной системы и реализации иммунологических процессов
| Этапы развития и функционирования клеток иммунной системы | События | Проявления апоптоза (в скобках — механизмы) |
| Формирование популяций лимфоцитов | Ранние этапы формирования популяций | Гибель избыточных клеток (вследствие дефицита факторов выживания) |
| Формирование антигенрас- познающих рецепторов | Выбраковка клеток с дефектами реаранжировки рецепторных генов (отсутствие сигналов от стромальных клеток) | |
| Селекция клонов | Гибель клеток, не распознающих аутологичные комплексы MHC—пептид (апоптоз «по умолчанию»), при положительной селекции и гибель аутоспецифических клеток (активационный апоптоз) при отрицательной селекции | |
| Дифференцировка субпопуляций | Апоптоз Т-клеток при несоответствии специфичности рецептора и корецептора | |
| Зрелые покоящиеся клетки | Гомеостатический контроль численности зрелых клеток | Гибель избыточных клеток (дефицит гомеостатических факторов выживания) |
| Элиминация старых клеток | Апоптоз старых клеток (вероятно включение эндогенной программы апоптоза) | |
| Иммунный ответ | Активация и пролиферация лимфоцитов | Активационный апоптоз (передача сигнала через мембранные рецепторы) |
| Созревание аффинности антител | Гибель низкоаффинных клонов В-клеток (конкуренция за антиген и помощь Т-хелперов) | |
| Элиминация эффекторных клеток | Апоптоз отработавших клеток (вероятно включение эндогенной программы апоптоза) | |
| Реализация цитотоксического эффекта лимфоцитов | Гибель клеток-мишеней цитотоксических лимфоцитов (передача апоптотического сигнала по перфорин/гран- зимному и рецепторному механизмам) |
Источник